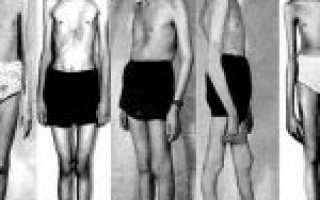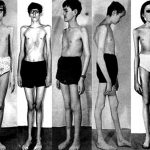
Синдром Марфана представляет собой генетическое заболевание, при котором резко себя проявляет недостаточность наследственного характера у практически всех соединительных тканей организма человека.
Недуг отличается различными проявлениями патологии скелетного, глазного или сердечно-сосудистого характера.
Сущность патологии
У подверженных этому синдрому людей могут появиться одно или несколько проявлений патолоии: гигантизм, эктазиозное поражение оболочки мозга, долихостеномелия, протрузия вертлужной впадины, арахнодактилия, плоскостопие, аневризма аорты, кифосколиоз, миопия, сильная деформация грудной области, эктопия глазного хрусталика. Диагноз устанавливается, базируясь, анамнезе, рентгенологии и исследовании генетики. Полностью от этого недуга избавиться невозможно, все лечение заключается в исправлении деформации и максимально возможном приближении к нормальному состоянию. Коррекция проходит над пораженными и деформированными системами человеческого организма.
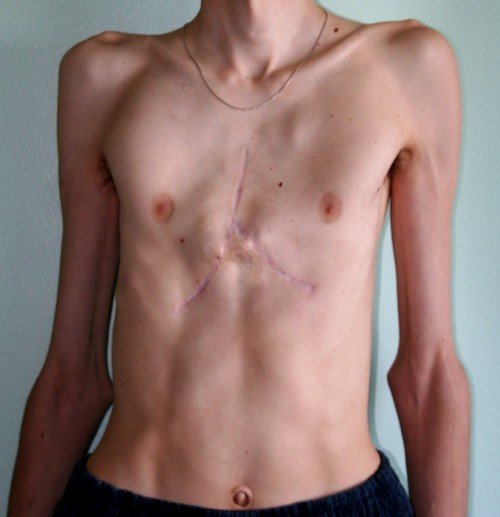
Синдром Марфана предполагает поражение коллагена, причем практически всегда эти поражения носят структурный характер. Кроме того, абсолютно во всех случаях вследствие появления и прогрессии синдрома он поражает либо одновременно, либо локально, одну из трех систем – сердечно-сосудистую, глазную или скелетную. Чтобы лучше понять такое заболевание, как синдрома Марфана, стоит посмотреть на фото, представленные в статье.
Кроме всего прочего можно сказать, что недуг является наиболее частым из всех редких синдромов генетического характера, поражающих коллагенную систему. Стоит отметить, что по статистическим данным, заболевание поражает 1 человека из 20 тыс. При этом не важны расовые и половые факторы.
Причины появления недуга
Вследствие того, что патология входит в генетический раздел заболеваний, ее причины стоит искать в генетики человека. По фото синдрома Марфана можно определить, что этот недуг педставляет собой именно генетическое заболевание.
Наследование заболевания происходит по доминантно-аутосомному характеру. При этом ярко выделяется высокопенетрантный и экспрессивный плейтропизм. Истоки заболевания начинаются с мутации одного человеческого гена, именуемого FBN1. Этот ген является крайне важным, так как благодаря ему синтезируется такое важное для организма вещества, как фибриллин, который по своей сути является белком структурного характера области межклеточного матрикса. Этот важный для нашего организма белок имеет в своей особенности важную функцию: он отвечает за сокращение соединительных тканей. Кроме того, он контролирует и эластичность этого рода тканей. Его дефицит приводит к тому, что соединительные ткани теряют эластичность, прочность и упругость. Кроме того, происходит сбой в структурах волокнистого характера. Все это приводит к тому, что больной не способен выносить любые физические нагрузки.
Существует более 1000 мутаций гена FBN1, синдром Марфана имеет множество проявлений. Различают как легкие формы недуга, которые сложно отличить от нормального состояния человека, так и тяжелые, которые крайне быстро прогрессируют и деформируют человеческое тело. Кроме того, мутация может присутствовать также и в других генах, что также влияет на появление множества разновидностей форм заболевания. Нелишним будет сказать, что практически в 80% случаев патология возникает в следствие генетической наследственности, и только в 20% появляется первичная мутация. Доподлинно не известны причины появления первичной мутации, но риск зачатия мужчиной ребенка, у которого в дальнейшем разовьется синдром Марфана, повышается с возрастом особенно после 35 лет.
В зависимости от поражения систем организма человека, недуг делиться на следующие разновидности:
- Стертая разновидность. Отличается тем, что поражение организма слабовыраженное, кроме того, недуг локализуется в одной или двух системах.
- Явно выраженная форма синдрома. К этой разновидности относят незначительные деформации в более чем 3 системах организма, явные деформации в любой системе организма, явные в более чем одной системе организма.
Лечение недуга
Стоит отметить, что одному врачу не под силу справиться с недугом, поэтому необхадима целая группа квалифицированных специалистов. К этой группе относятся следующие представители медицинского сообщества: терапевт, ортопед, генетик, кардиолог, хирург. В целом лечение сводиться к профилактическим мерам по предотвращению прогрессии заболевания и усугубления деформации организма. Под пристальным наблюдением должны находиться область и системы, связанные с сердцем человека. Кроме того, если поражены несколько систем, лечение всегда начинается с сердечно-сосудистой системы. Если отмечается диаметр аорты более чем 4 см, больному прописываются β-адреноблокаторы, а также ингибиторы или же антагонисты кальция. Операционное вмешательство необходимо при недостаточности клапанов сердца, явном расширении восходящей части и расслоении аорты, а также при пролапсе митрального клапана. Операция носит реконструированный характер и имеет весьма впечатлляющий процент продления жизни вплоть до 10 лет. Кроме того, в случае необходимости возможно протезирование митрального клапана. У беременных синдром Марфана требует срочного вынужденного деторождения, посредством проведения Кесарева сечения. В послеоперационный период в качестве профилактики прописываются антикоагулянтные и антибиотические медикаментозные препараты.
Коррекция зрения при данном синдроме проводится посредством подбора контактных линз или очков для зрения. В некоторых случаях понадобиться лазерная коррекция или лечение глаукомы, катаракты, а также возможна имплантация искусственного глазного хрусталика. Замена хрусталика глаза проводиться при сильном смещении естественного компонента. Если сильно выражены скелетные дефекты, проводиться хирургическая коррекция. Выполняется оперативная нормализация позвоночной области, протезирование суставов тазобедренной области или торакопластика. Кроме того, понадобиться проведении витаминной и метаболической терапии.
Прогноз течения заболевания устанавливается уровнем деформации и поражения систем организма. Отмечается быстрая прогрессия недуга и понижение качества жизни. В основном, больные редко доживают до возраста 45 лет. Своевременное обращение к специалистам и кардиокоррекция позволяют увеличить продолжительность жизни до 70 лет.